
近日,上海交通大学邹建新团队、比利时鲁汶大学Alexandru Vlad/林晓东团队设计了两个具有相似主链但π共轭程度不同的聚酰亚胺NUPI与PUPI,并系统研究了它们在含氯/无氯电解质中的电化学性能。结果表明:含氯电解质因其电化学稳定窗口窄、物种反应性强,会限制PI骨架的烯醇化程度与可逆性,从而限制容量;无氯电解质具有更宽电化学窗口与更温和的界面化学,可实现更深程度及更可逆的烯醇化,从而实现更高的可逆容量。此外,PI分子结构也起关键作用:相比π–π堆积较弱、微结构更无序的PUPI,NUPI能形成均匀的层状结构与更强π–π相互作用,有利于跨层电荷传输与Mg2+插入/脱出,因而表现出更优的储镁性能。此外,通过氧化石墨烯修饰隔膜进一步抑制了有机物种的溶解穿梭效应。原位/非原位表征与DFT计算揭示了烯醇化反应为核心的可逆快速储镁机制。综上,本研究阐明了PI分子结构与电解质化学在调控Mg2+传输与界面稳定性方面的协同作用,并建立了一种可推广的高性能镁金属电池协同设计策略。
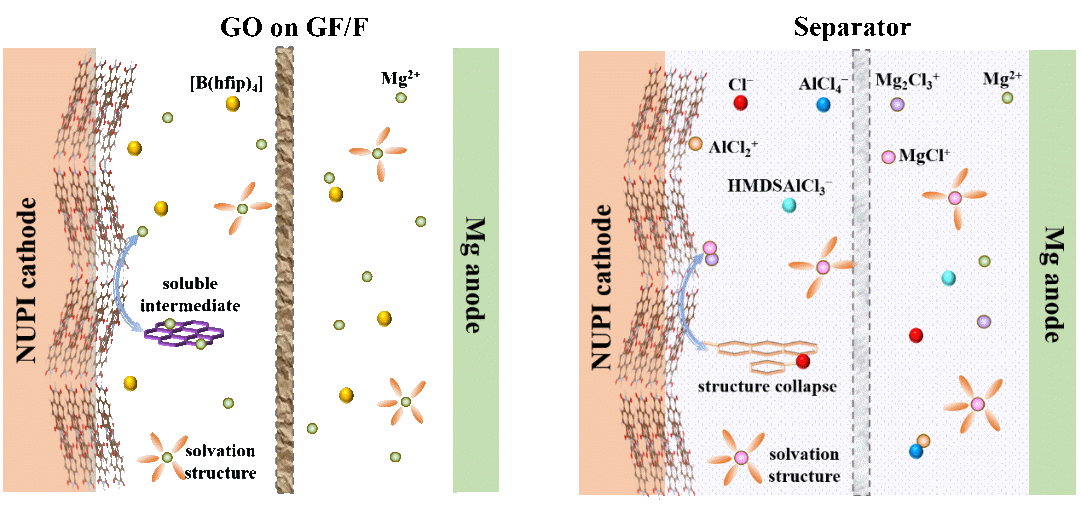
图1 文章TOC图
为阐明Mg2+储存机理,利用原位与非原位光谱测试。C=O在放电过程中逐渐减弱并最终消失,同时出现~1190 cm–1的[C–O–]2Mg2+伸缩新峰;充电后C=O恢复、[C–O–]2Mg2+减弱。NUPI于0.01 V下的[C–O–]2Mg2+信号更强,说明更深的放电使更多活性位点参与可逆烯醇化,而较窄电压窗口的电解液将不可避免地限制此过程。同步辐射原位FTIR进一步确认了其可逆性。如图2b 所示,在放电过程中,[C─O−]2Mg2+伸缩振动峰(约1190 cm−1)增强,而C═O(1680–1700 cm−1)和 C═C(约 1580 cm−1)伸缩振动峰的强度降低,这些变化在充电时会发生逆转。
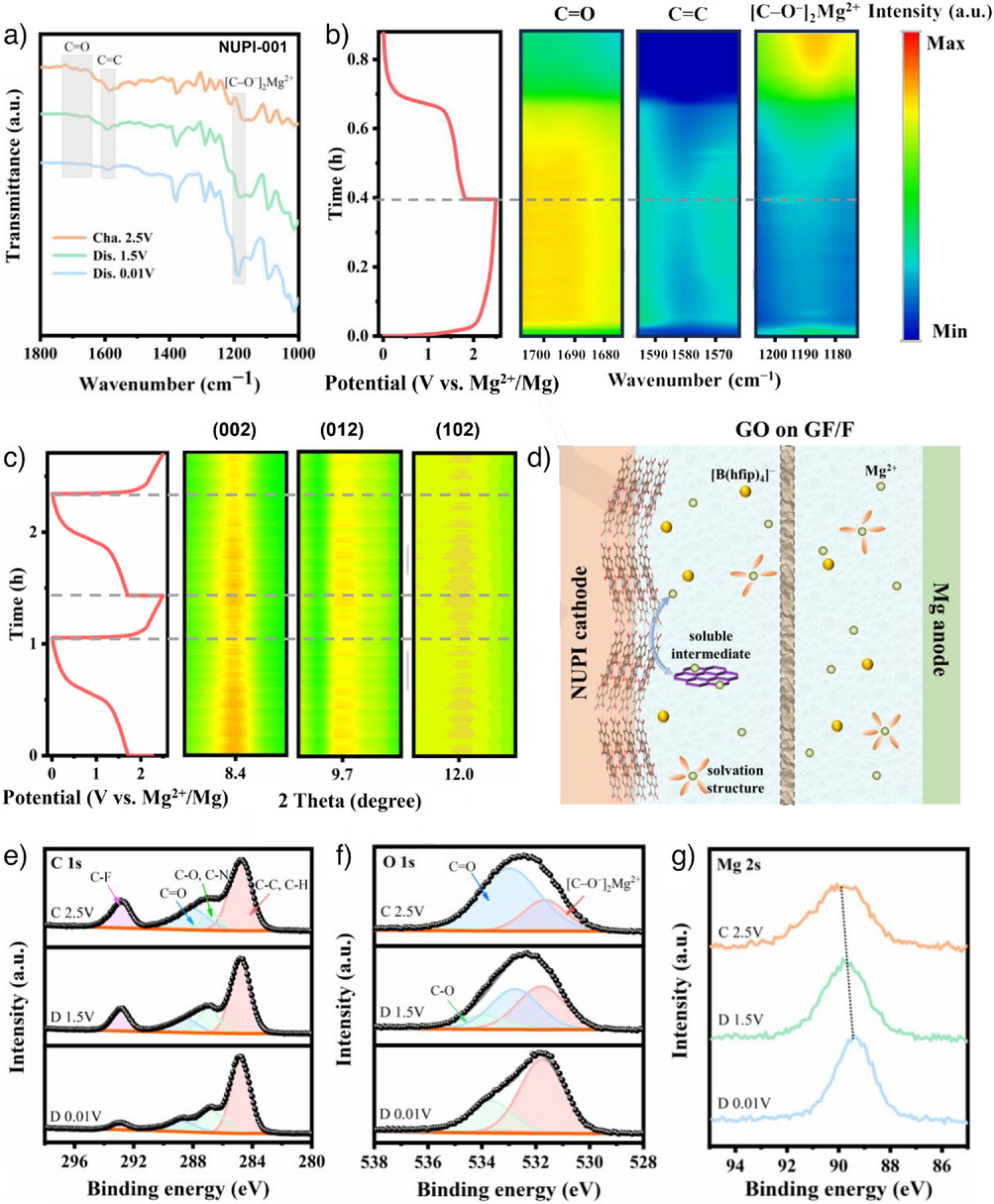
图2 聚酰亚胺基镁金属电池红外图(a:非原位;b:原位)
同步辐射原位XRD与非原位XRD显示在8.4°、9.7°、12.0°衍射峰在放电衰减、充电恢复,表明其高度可逆。π-堆叠有利于电子离域、降低界面电子积累、抑制电解液分解;无氯电解液能最大程度保持骨架有序性,使体系即便在深放电或高电压下仍具高可逆性。
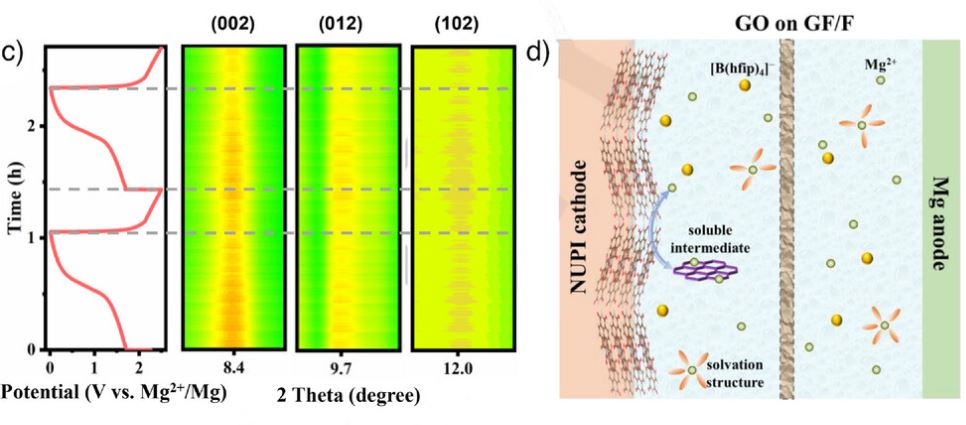
图3 同步辐射原位XRD图和镁存储示意图
国家蛋白质科学研究(上海)设施BL01B线站对上述工作的原位同步辐射红外光谱(in situ SR-IR)采集提供了技术支撑和机时支持。采用自制的原位红外电池装置(以BaF2片作为红外透射窗口)进行了原位 SR-IR 测试。为确保高质量的SR-IR 光谱,装置采用了反射模式,红外光垂直入射。通过平均 32 次扫描获得红外光谱。此前该课题组采用同样方法对CuS/PTCDA@450℃有机无机杂化正极进行了研究,以此提升硫化铜正极的镁离子存储性能(Adv. Funct. Mater. 2025, 35, 2413893.)。
文献链接:
X. He,X. Sun,R. Cheng,L. Hong,X. Lin,J. Li,P. Apostol,X. Xie,L. Shao,V. Frano,A. Vlad,J. Zou,Angew. Chem. Int. Ed. 2025,2522131.
https://doi.org/10.1002/anie.202522131
